

Why I bet on Lance
Over the years, I’ve learned the same systems lesson in a few different domains: when a data format no longer matches the way people actually use the data, every downstream layer starts compensating for it. Pipelines get more fragile, interactive workflows get harder, and performance problems show up in places that are easy to misdiagnose.
I first saw this in high-content microscopy. I saw it again in single-cell genomics. In both cases, the problem was a mismatch between the storage model and the workload that had evolved around it.
That mismatch matters more now than ever in biology. Single-cell atlases are larger, more multimodal, and increasingly expected to support repeated slicing, filtering, integration, and reuse across tools. These challenges extend beyond scale and cost to the architecture of data layouts and access-patterns.
That is the context in which I started building SLAF. This post is about why I bet SLAF's future on Lance, a storage format choice that ended up being foundational rather than incidental.
Biology's Long Tail of Formats
Biology has never had one canonical data format. It has had eras. Over two decades, each new technology has spawned its own storage format, each optimized for a different era's constraints.
Genomics birthed FASTA and FASTQ – text-based formats designed when a human genome cost $100 million to sequence, the world was lucky to have one genome, and the computational algorithms for aligning them to a reference were yet to be invented. Digital microscopy standardized around TIFF, a format that grew up in desktop publishing and photography, primarily concerned with preserving the best native resolution that imaging hardware could support, not with downstream computation. Likewise, single-cell transcriptomics standardized around H5AD, built on HDF5 groups storing non-zero entries of large sparse matrices to represent gene expression counts across thousands of cells.
These formats share a common origin story: they were designed for a world where data lived on local disk, computation happened in memory on a single machine, and the primary bottleneck was compute, not I/O. With the steady march of Moore's law driving up compute and memory capacity per dollar, that world no longer exists.
Today, datasets live in object storage, and computation runs across distributed clusters. The bottleneck is no longer whether we can compute, but how efficiently we can move data into compute.
From TIFF Bottlenecks to Cloud-Native Thinking
Years before SLAF, I was working on a neuroscience drug discovery platform at Herophilus, where we developed brain organoids (industrialized mini-brain models) to discover novel medicines for brain disease. Organoids serve as the next best counterfactuals to (hard to obtain) post-mortem patient brain tissue. In a world where it takes a decade to assemble a 10k sample size post-mortem brain tissue biobank, our organoid factory produced 50k mini brains per week. To study their detailed emergent biology, we put them in micro wells and imaged them. Our automated imaging work cells generated high-resolution microscopy at scale: spanning multiple z-stacks, multiple timepoints, multiple fluorescent channels across multiple slides or well plates. We schlepped TIFF files to the cloud, indexed the metadata, and built a data lake to serve diverse workloads.
Data scientists explored datasets in notebooks. Data engineers built batch pipelines and ran them on spot instances. ML engineers trained vision models and deployed inference systems. And everyone wanted on-demand visualization. Agents were not yet part of the stack, but if they had been, they would have touched all of these workloads too.
TIFF hit its limits quickly. It worked by copying gigabytes at a time to developer nodes or spot instances. We accepted this as the cost of doing work, but we struggled for random, concurrent, low-latency access patterns like those in visualization. So, we forked the format. We built internal JPEG-based image pyramid copies and served them with tile servers to browsers. When we finally migrated to zarr, a cloud-native chunked storage format for n-dimensional arrays, we saw the same pattern many teams discover: once data is chunk-addressable and remotely readable, whole classes of workflows become practical. We streamed imaging slices to distributed compute for embarrassingly parallel workloads that scaled both vertically (across cores) and horizontally (across machines), helping us realize many storage, latency, throughput and cost-savings multiples across all of them.
That lesson stayed with me.
At Herophilus, we also did single-cell transcriptomics on our organoids, without adopting cloud-native storage. It felt "fine" as late as 2022 because datasets were still manageable with local workflows. Then, the scale changed.
The Single-Cell Inflection Point
The average adult has 37 trillion cells, each carrying the 3 billion base pair codebook of the human genome in its nucleus. What makes a cell unique is the genes that are expressed over its lifetime. The ability to measure gene expression (RNA molecules) and proteins in single cells and map their spatial distribution has ushered in unprecedented detail in understanding molecular signatures of cell types and tissue composition, developmental biology: how cells differentiate from stem cells, cancer biology: how cancer cells evade drugs and immune systems, and cell-cell interactions.
Here’s the high-level journey that RNA molecules in our cells make on their way to a count matrix: in droplet-based single-cell RNA-seq, we suspend cells in droplets so that each droplet captures one cell plus a bead coated with large molecules of nucleotides (oligos) – cell barcodes determining which droplet/cell this came from, a UMI (a molecule tag so duplicates from the polymerase chain reaction (PCR) can be collapsed), and a poly‑T tail that grabs mRNA. Then we deliberately pool everything: all droplets, often many samples multiplexed together, go through one sequencing run. The sequencer outputs short reads; we align (or pseudo align) them to a reference transcriptome/genome, decode the barcodes/UMIs, and count molecules per gene per cell. The end product is the canonical artifact of the field: a huge, extremely sparse count matrix (cells × genes), plus metadata telling you which biological sample or condition each cell belongs to and QC attributes.
Single-cell datasets have grown 2,000-fold in less than a decade. What used to be 50,000 cells per study as late as 2022 is now 100 million cells in a single release. In February 2025, Tahoe Therapeutics released Tahoe-100M: 100 million transcriptomic profiles measuring drug perturbations across 50 cancer cell lines. It's the world's largest single-cell dataset, 50 times larger than all previously available drug-perturbation data combined. They released it on Hugging Face with this extremely quotable figure.
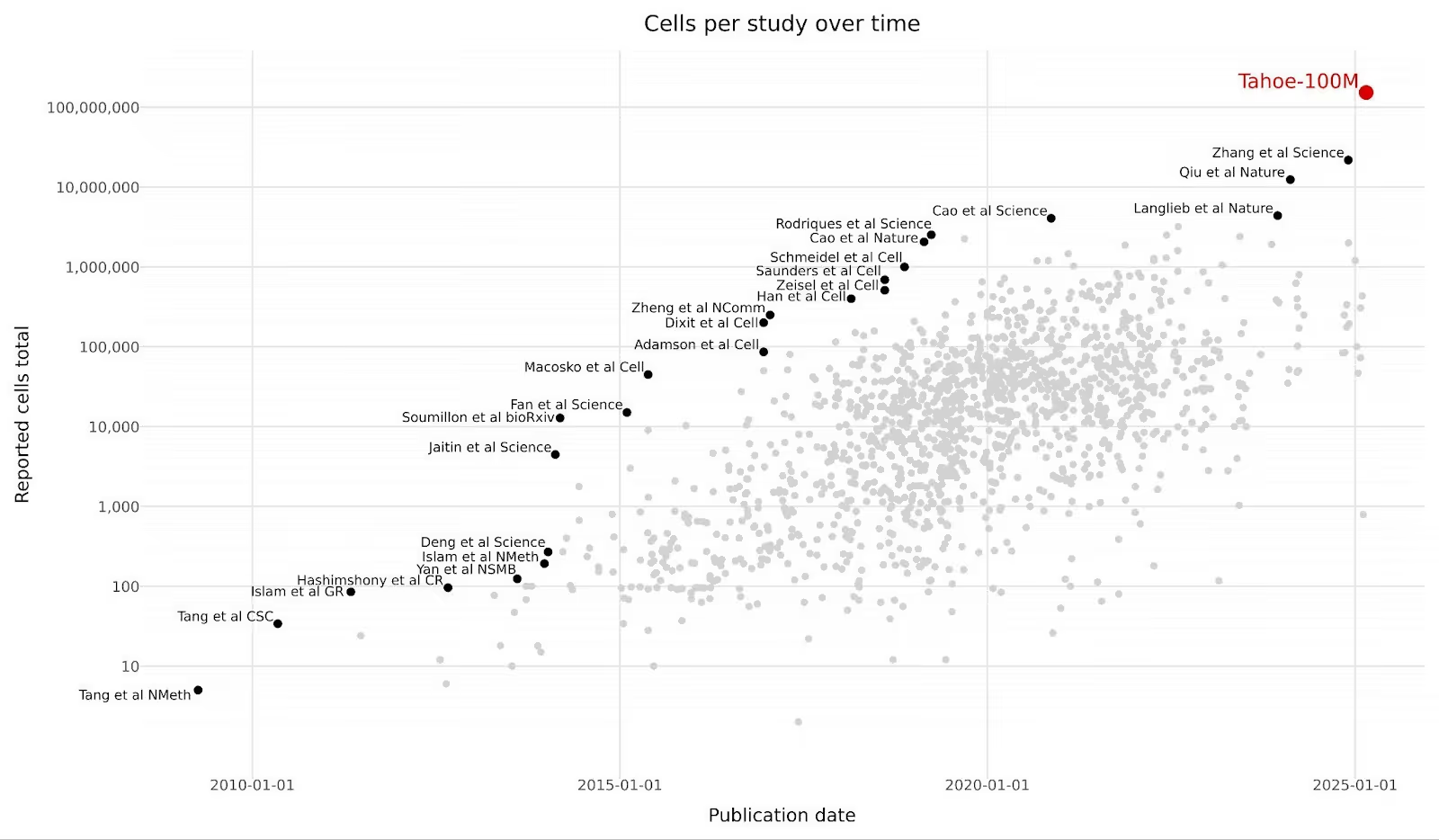
With Illumina announcing a billion cell atlas project in January 2026, the scaling continues at a rapid pace.
If you're curious about what has driven such scaling, technological innovations in library preparation (transforming cell-specific RNA molecules to barcoded droplets; see 10x Chromium overview), sequencing (converting RNA fragments to raw base pair reads; see Illumina’s sequencing-by-synthesis overview), and demultiplexing (computationally reverse engineering sample identity from multiplexed sequencing via genotypes; see demuxalot), along with healthy market competition (see Alex Dickinson's commentary on the evolving competitive dynamics in sequencing cost per cent), have conspired to make a new Carlson curve for single-cell biology.
From a history-of-ML perspective, this is also the bitter lesson playing out again: qualitative leaps rarely come from clever architectures alone; they come when the dataset gets big enough that general-purpose learning finally has room to work. The recurring pattern is new dataset release + a model that can actually absorb it = a new era:
The open question now is whether we’re watching the same movie happen in biology: Tahoe‑100M (and what comes after it) + single-cell foundation models = the “virtual cell” era, where representation learning over atlases becomes as foundational as alignment or clustering.
What does this ambition translate to in terms of computational appetite? I see two patterns.
- Existing local-disk workflows become operationally expensive.
- New workloads (foundation model training, embedding retrieval, atlas distribution) require capabilities the old formats did not prioritize.
What Breaks in the Status Quo
Let’s take a deeper dive into a canonical workload using status quo tools.
Specifically, the status quo workflow for single-cell data looks something like this: download terabytes from a public bucket to your own bucket, then to local storage, load or stream it into memory, transform it into library-specific in-memory formats. Limitations surface in the form of various ceilings: egress costs, network throughput, local storage capacity, finite RAM per node, as well as various design decisions that leave gains on the table: single-threaded I/O, single-threaded compute, underutilized CPU cores, overprovisioned GPU clusters.
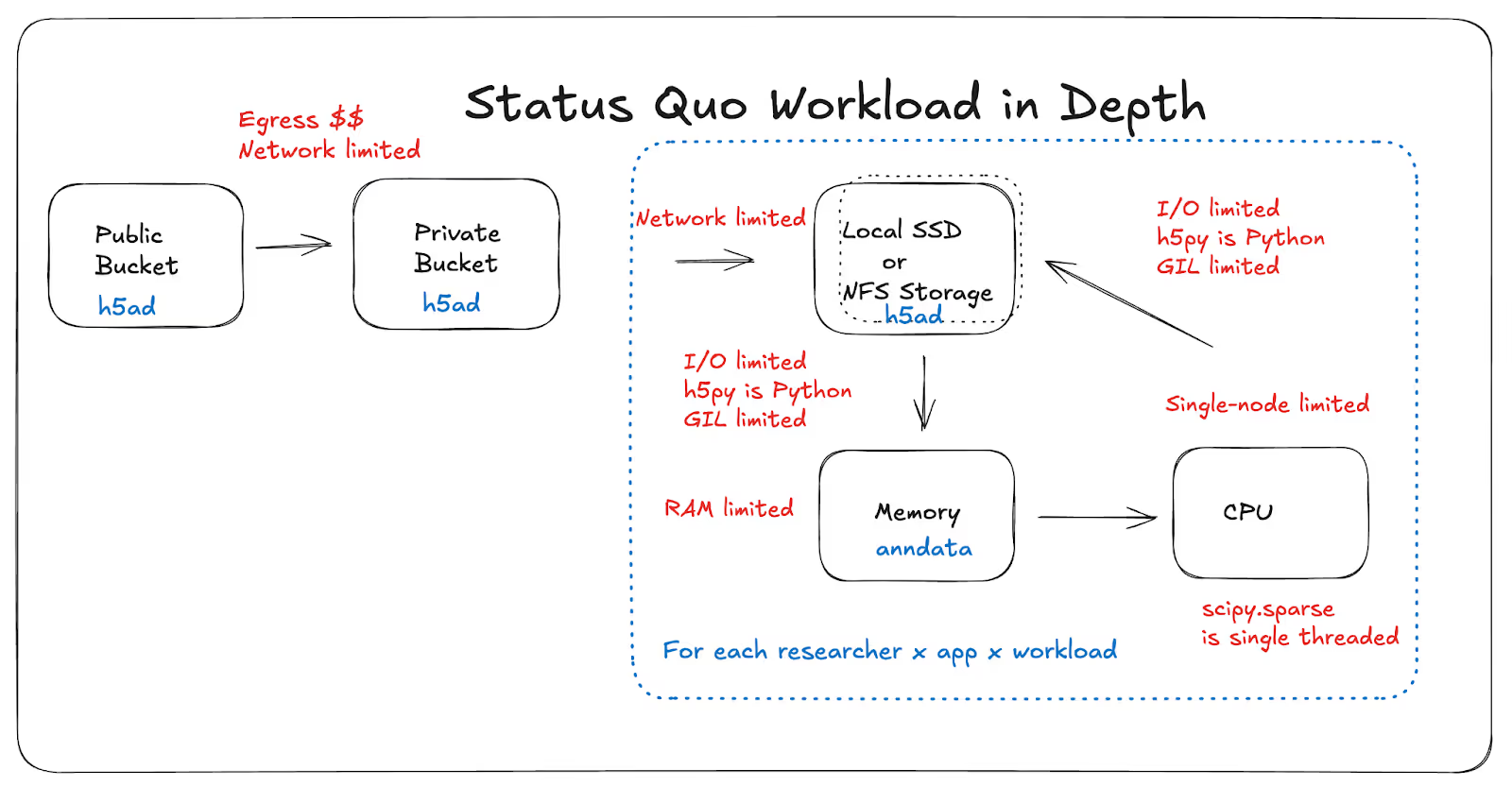
H5AD (HDF5-backed sparse arrays) remains a great format for many local analysis workflows. But at atlas scale and in multi-user/cloud scenarios, failure modes pile up:
- Read amplification problems: Large transfers become a prerequisite for querying even small slices.
- Egress costs: Teams serving large atlas datasets are paying down egress costs even for random access, compounded by read amplification issues
- Data lineage problems: Teams duplicate entire files per user/job, modify them in-place and spaghettify data lineage.
- Network bottlenecks: Streaming to compute just isn't fast enough, warranting colocated copies of large training corpora near GPU clusters, increasing storage and transfer costs
- I/O bottlenecks: Reading hdf5 groups into Python via `h5py` is GIL-limited and does not provide true concurrency.
- Compute bottlenecks: Manipulating `scipy.sparse` objects in memory is single threaded and wastes ubiquitous, cheap, modern multi-core CPUs.
- GPU costs: As I've written about before, modern GPUs are voracious and modern neural networks are impressionable. They need batches arriving before they have completed a training step in a randomized order. With the wrong storage format, i/o bottleneck, decompression algorithms, or shuffling methods, GPUs can remain idle 90% of their uptime in between useful work(the training step).
- Latency problems: Data visualization dashboards and agents, may require only a sliver of datasets (subset of cells or genes) and are slowed down by huge up-front RAM needs for web services (non-starter) or slow random access from disk (non-finisher).
The issue is not that H5AD is bad. The issue is mismatch: the format was designed for a different center of gravity than object-store-native, concurrent, streaming, analytical + exploratory + ML workloads.
Two Problems of Different Scale
Framing it this way, I found it useful to separate scale issues into two classes.
Class 1: Existing workloads get harder
PCA/UMAP/QC pipelines, interactive notebooks, and collaborator sharing still exist – they just become operationally fragile and expensive with tens of millions of cells. I've heard from pharma leaders that their time is spent petitioning for budgets to spin up 2TB RAM instances for exploratory, interactive, or analytical work.
Class 2: Entirely new workloads appear
- Atlas-scale open data distribution
- Foundation model pretraining/fine-tuning
- Large embedding stores and ANN search
- Agentic query workflows over biological metadata + expression
The ecosystem's response has been fragmentation: one format for one workload, another for another. That is understandable, and harkens back to what we did at Herophilus for microscopy, but it forks teams, and becomes expensive to maintain.
The Core SLAF Vision
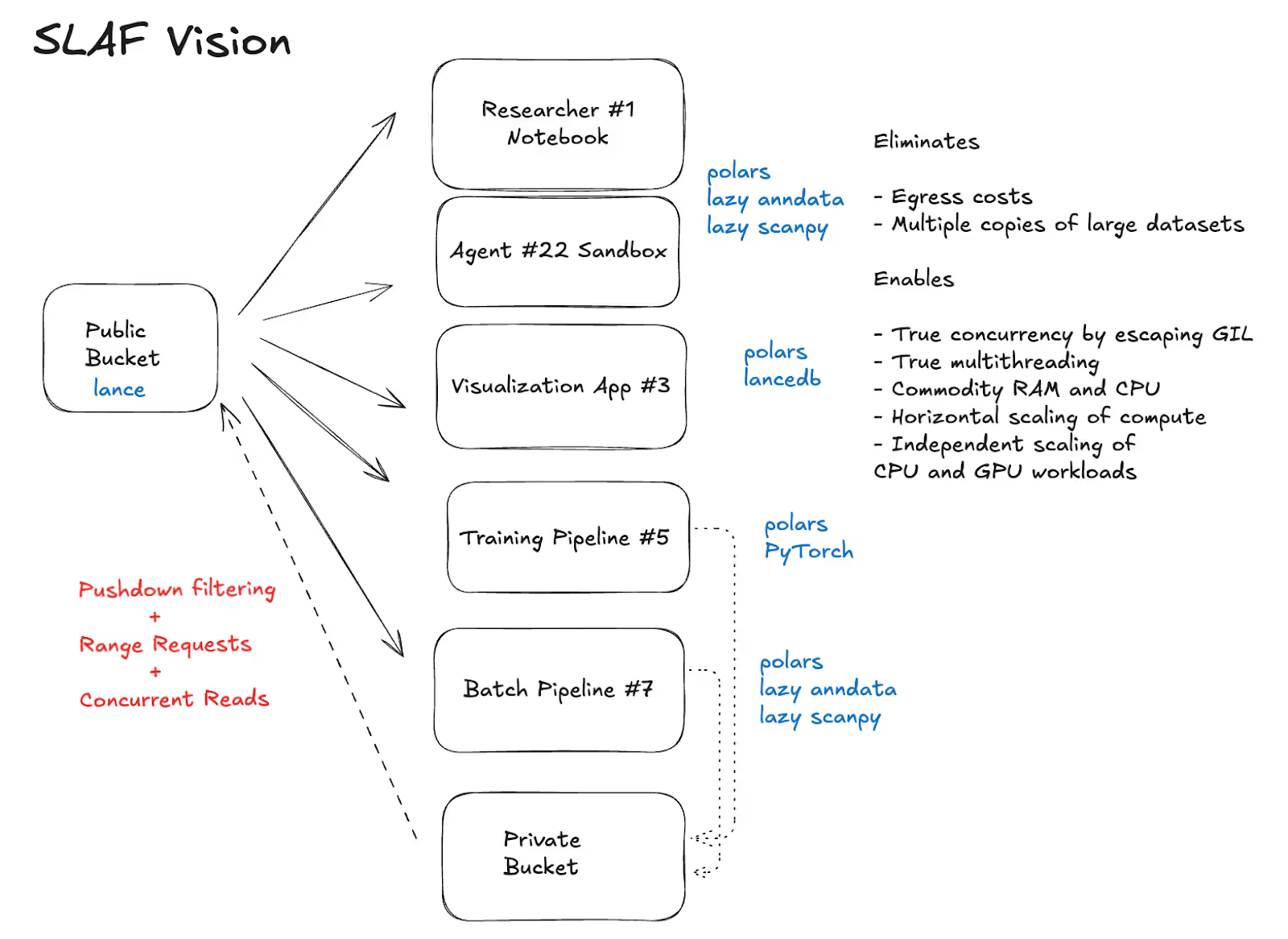
SLAF’s “X for Y” vision is “Databricks for single cell-omics”. Store once in a data lake, and stream to every imaginable workload. Instead of the status quo canonical workload, we want to see streaming support for exploratory work, interactive visualization, analytical batch jobs, foundation model training, batch and on-demand inference, all while managing data evolution as a first class citizen throughout this lifecycle.
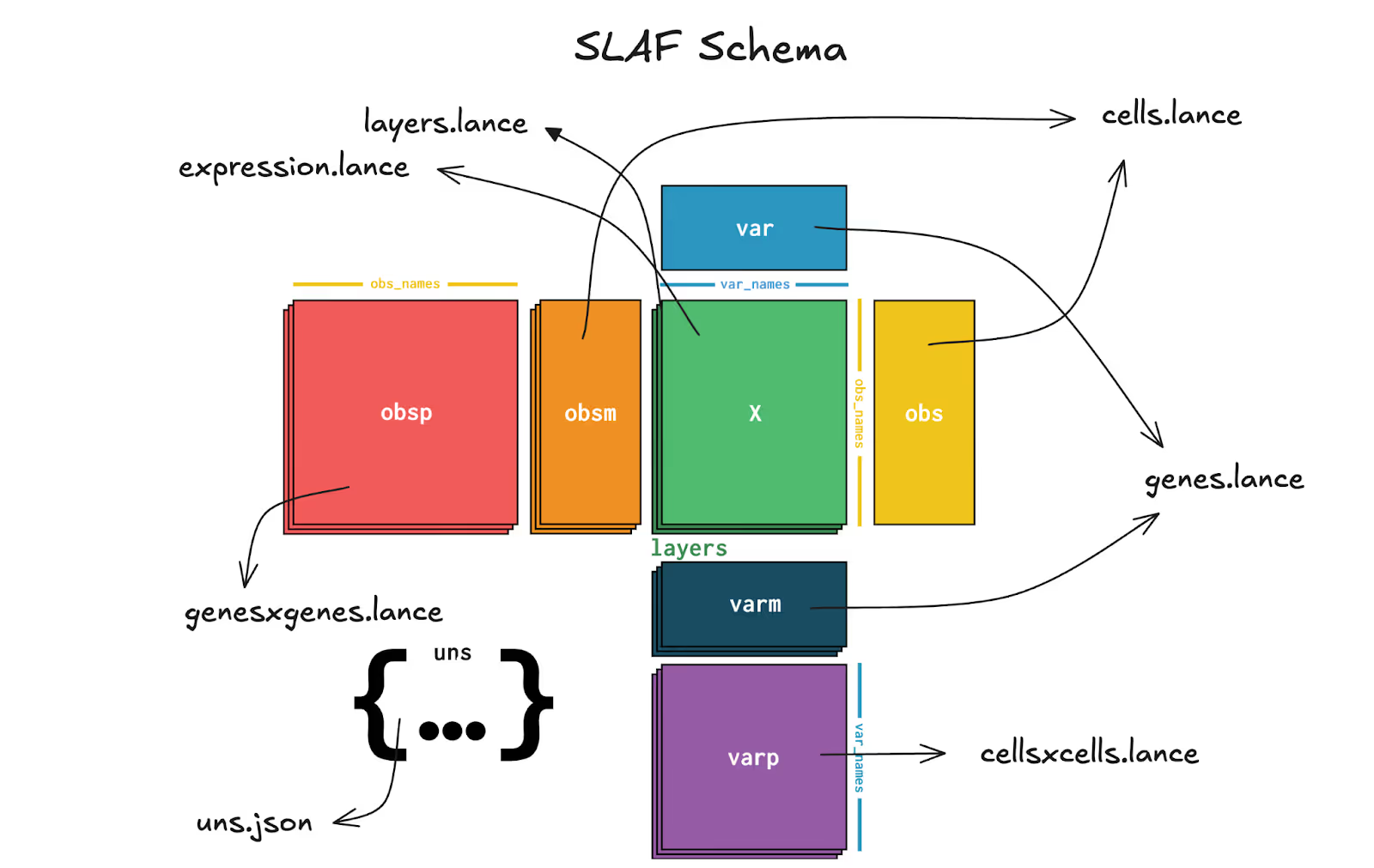
SLAF's first major decision was simple but consequential. Represent expression as sparse COO-like records in Lance. Conceptually:
expression:(cell_id, gene_id, value)recordscells: cell metadata tablegenes: gene metadata table
There are a few additional wrinkles that complete the anndata schema, the open data model for expression and metadata on which single cell omics operates. I won't get into it here, but as I've written about before, this design turns expression into a table-native representation that benefits from all the hard fought victories of the OLAP movement over the past decade: columnar compression, bloom filters, shard-aware query plans, pushdown and projection pre-filtering, optimized joins, and streaming batch reads.
The implications for large sparse single cell count data are key: store only non-zero observations, keep metadata queryable in-column, and read only what a workload actually needs.
With this schema in place, there were decisions to be made about where Lance as a table format fit into a disaggregated stack for maximum modularity.

At the bottom, users pick a storage layer: typically object store, but with Lance x Hugging Face integration, a bonus option is to store SLAF data in a Hugging Face datasets repo, as I have done for a few bellwether single-cell Atlas datasets in the field. In fact, streaming from Lance tables on Hugging Face can have ~1.5x higher throughput (because of async i/o prefetching) than the native datasets library.
Next comes the choice of a table format.
Why Lance Specifically?
When I evaluated modern formats that are already cloud-native and shard-aware (Parquet + Iceberg, TileDB, Zarr, Vortex), most of them were very strong on one or two axes. But SLAF needed the full bundle below, in roughly this order:
- Cloud-native storage: the same dataset should work locally and on object storage without a bespoke serving layer.
- Fast random access: both analytics-style scans and “give me these specific rows/cells” should be cheap.
- ACID-like correctness properties: multiple writers, append-heavy workflows, and a path to safe evolution.
- Time travel and versioning: atlases evolve; users need stable snapshots and reproducibility.
- Vector-native types and indexing: embeddings aren’t an add-on anymore - they’re a primary artifact.
- Separation of concerns: I want to mix-and-match query engines without rewriting storage or sacrificing pushdown.
- Manifest-based versioning without 100x copies: update metadata and schemas without rewriting the world.
- Re-indexing without rewrites: add or rebuild indexes as data and workloads evolve.
- Advanced sharding / compaction via Python API: control the physical layout as a first-class lever.
- Hugging Face integration: distribution is a workload; make it frictionless.
Lance was the first format I encountered where I didn’t have to pick which workloads to compromise on.

How Lance Became Operational Across Workloads
Once I settled on the table format, the next step was making sure workloads were performant with the right query engine and the right user interfaces for every workload.
Atlas distribution and ad-hoc analytics
At atlas scale, “download the dataset, then query it” is a failure mode. A useful format needs: concurrent streaming reads over object storage, column projection (ship only what you ask for), predicate pushdown (skip irrelevant data early), and evolution/versioning that doesn’t imply full rewrites. Here is where interoperability with query engines becomes critical.
A full discussion of query engine choice is beyond the scope of this post, but at a high level, we have amazing options with OLAP heavyweights like DuckDB, DataFusion and Polars. While I explored each of them and found Lance interoperability to be strong (and getting stronger), I settled on Polars because my workloads were compatible with several DataFrame-like operations downstream, and I’m excited about the ability to accelerate query processing on GPUs with Polars.
In benchmarks that you can reproduce (local disk and CPU only so far), we’re seeing 10-100x acceleration in metadata queries and similar fold improvements in memory efficiency for expression queries.
Batch processing pipelines
Large biological jobs are often embarrassingly parallel if your storage unit aligns with your compute shard. With Lance, fragment-oriented reads enable straightforward work partitioning by assigning fragments (or fragment ranges) to workers, streaming Arrow batches within fragments, and keeping ephemeral compute close to object storage.
Zarr shines when the primitive is an N-D array chunk, but single-cell workloads are sparse arrays and chunks of CSR schema without linked metadata doesn't feel complete for workloads that require “filter, join, aggregate, and then slice.” operations. That ability to store metadata next to gene expression and exploit the full strength of the OLAP movement felt substantially more accessible in Lance.
Foundation model training data loaders
With increasingly powerful GPUs, the training bottleneck is frequently not model architecture, it’s data delivery.
The old pattern results in multiple full copies: a canonical scientific file, a training-optimized derivative, temporary job-local copies for every training experiment’s slightly bespoke need to see batches in a slightly different way.
Lance’s fragment layout and Arrow-native batch streaming make it natural to build shard-aware prefetchers that keep GPUs fed directly from the lake artifact. A tight integration with Polars make it seamless to build custom on-the-fly preprocessing, tokenization, and shuffling operations (SLAF’s dataloaders achieve randomness while remaining blazing fast) that push down filtering to the storage layer, push preprocessing and tokenization into vectorized ops on the CPU and deliver random, tokenized batches with zero inter-training step latency.
Here, it is worth noting that most modern cloud-native formats are “streamable” in the trivial sense. The differentiator for training is whether streaming is prefetch-first: Lance’s async I/O and prefetching under the hood make shard-aware dataloader prefetchers trivial to build (see SLAF’s dataloader design), whereas many other formats leave users reinventing the I/O scheduling layer (and its performance traps). In verifiable benchmarks, we’re seeing that SLAF dataloaders reach up to 100x the throughput of status quo dataloaders based on the h5ad format.
Inference and embedding management
Single-cell pipelines are increasingly embedding-first. You need to write embedding vectors at scale, often from distributed workers concurrently. When you augment an atlas with new data, you need to update the index. At serving time, you need to query nearest neighbors quickly without maintenance windows, and with pre and post filtering on cell metadata and expression-derived features. Silo-ing this workload to a vector database and maintaining live links between for this workload feels like an overwhelming project for so many practitioners, that even though science has started to be informed by cell and gene embeddings, we haven’t seen the storage layer catch up.
Formats like Vortex are starting to move in this direction, but storing a vector column with SOTA compression isn’t the same as a table format with ACID-like semantics and easily updated vector indexes. Treating vectors as first-class columns with native indexing is the difference between “I can store and query embeddings” vs. “I can build retrieval-native biology workflows.”
Interactive visualization
Visualization rarely needs the full expression payload. It needs selective projections (coordinates + labels + small metadata windows) with low latency. Formats optimized for bulk scans can still be painful for interactive sampling if random access and nearest-neighbor lookups aren’t first-class design constraints. In practice, a good web experience often means predictive prefetch: as a user hovers/filters/zooms, the service prefetches the points they’re likely to click next via ANN search at interactive speeds. That loop only works if your storage layer supports fast random access and vector indexes.
Closing Thoughts
Familiar Interfaces, Bleeding-Edge Internals
The one trait shared by all software I’ve loved building and using is that most users should not have to care about how it works. Given that the humans behind heterogeneous workloads of SLAF are just as heterogeneous, user experience is top of mind for me, and often trumps workload efficiency.
In practice, this led me to build two very different interfaces on top of Lance and Polars.
The first are a set of lazy views into Lance expression and metadata tables that allow array-like slicing and DataFrame-like filtering, while hiding the sophisticated under the bleeding edge OLAP engines that push down to Lance. The result is that computational biologists are able to write code that resembles their existing workflows in their library of choice (anndata and scanpy).
The second is a dataloader class as a generator that behaves like PyTorch’s DataLoader, while hiding multithreaded async prefetching over Lance’s native prefetching of fragments, and Polars-based preprocessing on top of zero-copy Arrow batches. The result is that ML engineers training foundation models should write training loops as before and forget about migrating copies of their training data to GPU clusters.
This is how infrastructure wins: by disappearing behind stable interfaces.
What Comes Next
If you’re a systems researcher or builder: I’m excited that you got to learn about a new data modality and its unique challenges. I hope this will inspire you to bring your skills to biology and science.
If you’re a single-cell computational biologist or ML engineer marching into a new era of scale, you’ve found a next generation tool for all your workloads. Give it a try!
If you’re building agents, then having them execute complex workflows in near real time by streaming from object store is a natural next step and makes your agents less resource hungry than alternatives. I encourage you to lean in and host your agent’s datasets on object store or Hugging Face, backed by SLAF or Lance.
The roadmap from here is less about one benchmark and more about ecosystem shape, moving towards robust multi-node training integrations, production-grade embedding lifecycle management, deeper GPU-accelerated query/DataFrame paths, support for data formats being single cell transcriptomics, and cleaner interoperability bridges for existing single-cell tools. The larger goal continues to drive towards one format contract that keeps biology data usable as scale continues to grow.
SLAF is an MIT-licensed open source package and welcomes community contributions.
What This Means for Teams Adopting Lance
I want atlas-scale biology to be broadly usable, and not just by teams with dedicated infrastructure engineers. To get there, we need storage choices that make the common path fast, composable, and cloud-native by default.
For SLAF, betting on Lance was that choice. I chose it because I needed a foundation that could survive workload divergence, without forcing format fragmentation or constant data duplication. In practice, Lance was the first format where the architecture felt like the future instead of a patch over the past.
Resources
Github: https://github.com/slaf-project/slaf
Docs: https://slaf-project.github.io/slaf/
Hugging Face: https://huggingface.co/slaf-project
Blog: https://slaf-project.github.io/slaf/blog/
Appendix
This section addresses common questions around "why not Parquet / Iceberg / Zarr / TileDB / Vortex?"
Why not Parquet for everything?
Parquet is fantastic for columnar scans, compression, and interoperability. The pain starts when you need both high-throughput scans and fast random access patterns, while also evolving the dataset (indexes, versions, schema) in a way that stays ergonomic for iterative ML + analytics + interactive use.
Why not Iceberg (with Parquet) for table semantics?
Iceberg is a strong table layer, but SLAF also needs a storage artifact that is shard-aware and stream-friendly for ML data loading and for interactive sampling, not just “queryable by engines that speak Iceberg.” The question isn’t whether Iceberg works – it’s whether the full stack stays simple when you add embeddings, ANN search, and training throughput as first-class requirements.
Why not Zarr for chunked access?
Zarr is great when the core object is an N-D array and your access is mostly chunk slicing. SLAF’s core object is a sparse observation table + rich metadata + embeddings, and the workloads are dominated by filter/join/aggregate patterns where table semantics and pushdown matter as much as chunking.
Why not TileDB?
TileDB is a serious system, and it does have table capabilities. For SLAF, the differentiators ended up being table-format ergonomics at atlas scale: append/evolve without proliferating copies, plus fine-grained, Python-level control over sharding/compaction, while keeping random access, versioning, and vector-native behavior on the same contract.
Why not Vortex (or scan-optimized formats) for analytics?
Scan performance is necessary but not sufficient. SLAF has to serve analytics, ML training, interactive exploration, and embedding retrieval from the same canonical artifact. Vortex is compelling on compression and scan speed, but SLAF also needs table-format semantics (ACID-like evolution + time travel) and vector indexing as first-class features.



